
دانشجویان و پزشکان، عمدتاً از وارد شدن به مباحث لوسمی حاد (Acute Leukemia) پرهیز میکنند. تعداد بیشماری دارو با اسمهای جدید، تعداد زیادتری جهش با اسمهای غریب، یادآوری خاطرات دردناک از دوران تحصیل از این بیماران، گیجکننده بودن تقسیمبندیها و این فکر که لوسمیها برای هماتولوژیست است، دانشجویان را از این مبحث دور میکند.
مدرسهی پزشکی برای درک بهتر این دروس، درسهایی در آینده آماده خواهد کرد. اما در میان همهی انواع سرطانهای مغز استخوان، Acute Promyelocytic Leukemia که همان Acute Myeloid Leukemia, M3 است، جایگاه ویژهای دارد. چرا که شناختن AML – M3، شک کردن به آن در مواقع لازم و شروع درمان اورژانسی است که میتواند یک بیماری صددرصد کشنده را به یک بیماری که علاج (Cure) دارد، تبدیل کند.
چرا دو اسم؟ APL صحیح است یا AML-M3؟
یک سیستم طبقهبندی قدیمیتر به نام FAB وجود دارد که مخفف French-American-British است. در FAB انواع AML بر اساس شکل سلول (مورفولوژی) تقسیمبندی میشدند و این نوع از AML نام AML-M3 را به خود اختصاص داد.
این ترمینولوژی در دستهبندی جدیدتر که توسط سازمان جهانی بهداشت (WHO) انجام شده و بر اساس جهش ژنتیکی است، به صورت Acute promyelocytic leukemia with PML RARA نامگذاری میشود.
هیچ کدام غلط نیستند؛ اما هنوز هم در جامعهی پزشکی ایران عبارت AML-M3 رایجتر است. هرچند APL شیواتر است و به ما اطلاعاتی در مورد پاتوفیزیولوژی نیز میدهد.
اپیدمیولوژی و پروگنوز (پیشآگهی)
APL بدخیمترین نوع AML است و بدون درمان بیشک کشنده است و بیماران به صورت میانگین بیشتر از یک ماه زنده نمیمانند.
APL تفاوتهای مهم و کلیدی با دیگر انواع AML دارد. عمدهی ابتلا به AML در همه انواع آن در سن بالای ۴۰ سال است، اما برای APL زیر ۴۰ سال است.
این نوع علاجپذیر (Curable) است. انواع دیگر AML در هر لحظه امکان عود (Relapse) دارند و ممکن است وضعیت بیمار وخیم شود. به صورت کلی میزان زندهمانیِ (Survival) پنجساله در انواع دیگر AML کمتر از ۳۰ درصد است؛ اما با رویکردهای درمانی فعلی حدود ۸۵٪ بیماران APL پیشآگهی بلندمدت بسیار خوبی دارند و بیماریشان علاج میشود.
پاتوفیزیولوژی در APL
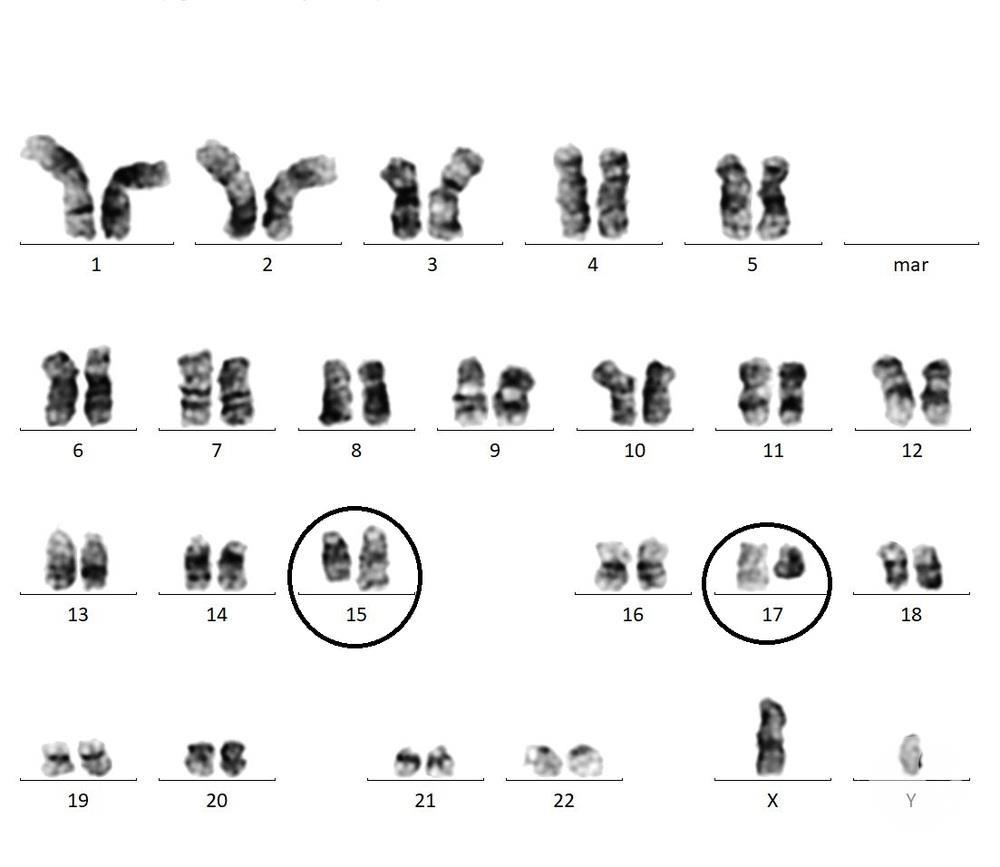
یک جابهجایی کروموزومی (Translocation) بین کروموزومهای ۱۵ و ۱۷ اتفاق میافتد. در این جابهجایی دو ژن PML و RAR-α کنار هم قرار میگیرند. این اتفاق، هم در درمان برای ما اهمیت دارد و هم در تشخیص.
t(15;17) –> PML-RAR-α fusion gene
برای تشخیص همین ژن را با روشهایی مثل PCR مییابیم و برای درمان نیز همین محصول RAR-α را هدف میگیریم که یک گیرنده برای رتینوئیک اسید است.
بیماران مبتلا به AML با چه علائمی میآیند؟ در آزمایشها انتظار چه چیزی را داریم؟
بیماران مبتلا به AML به صورت کلی با شکایات و علائم ناشی از کاهش هر سه رده سلولی مغز استخوان (پانسیتوپنی) میآیند. میدانید که پیشوند پان (Pan) به معنای همه است و سیت (Cyte) از سلول میآید و پنی از Penia که به معنای کاهش است. حدود ۸۵٪ بیماران AML-M3 در بررسی اولیه پانسیتوپنی دارند. بقیه انواع AML معمولاً با لکوسیتوز به همراه آنمی و ترومبوسیتوپنی مراجعه میکنند.
آنمی، نوتروپنی و ترومبوسیتوپنی خود را به شکل ضعف و خستگی زودرس، عفونت مکرر یا شدید و خونریزی لثه، اپیستاکسی، اکیموز و منوراژی نشان میدهند. معمولاً ترکیبی از این شکایات وجود دارد. در دیگر انواع AML معمولاً علت این خونریزی، کاهش پلاکتهاست. اما ویژگی منحصر به فرد APL، خونریزی ثانویه به DIC است.
DIC که مخفف Disseminated Intravascular Coagulation است، به علت گرانولهای پرومیلوسیت ایجاد میشود. این گرانولها همانند فاکتور بافتی، مسیر انعقادی را فعال کرده و در نتیجه فاکتورهای انعقادی مصرف میشوند. در نتیجهی فعال شدن مسیر انعقادی فیبرینوژن به فیبرین تبدیل شده و این تورهای فیبرینی در داخل عروق، گلبولهای قرمز را به دام میاندازند و باعث بسته شدن عروق میشوند (ترومبوز).
همچنین در برخورد گلبول قرمز با این رشتههای فیبرینی، گلبول قرمز میشکند و تکههای گلبول قرمز (شیستوسیت) درست میشود.
به خاطر مصرف شدن فاکتورهای انعقادی، تستهای انعقادی در APL تقریباً همیشه مختل است. یعنی تقریباً همیشه PT، PTT و INR مختل بوده، سطح Fibrinogen کمتر از ۱۵۰ میلیگرم در دسیلیتر و سطح D-Dimer بالای ۱۰۰۰۰ نانوگرم در دسیلیتر است. پس اگر شک کردیم که AML-M3 است و تستهای انعقادی نرمال بود، تشخیصمان زیر سؤال میرود.
چه زمانی به وجود AML-M3 شک کنیم؟
هر زمان که پانسیتوپنی در همراهی با خونریزی وجود داشت یا هر زمان که پانسیتوپنی به همراه شواهد ترومبوز وجود داشت (مثلاً شواهد بالینی یا پاراکلینیکی به نفع Cerebrovascular Accident یا Deep Vein Thrombosis).
دو نمونهی کلاسیک و شایع APL به صورت زیر است: خانم یا آقایی با سن زیر ۳۰ سال که به دنبال کشیدن دندان، خونریزی از محل دندانش قطع نشده و در بررسیها متوجه پانسیتوپنی میشویم. یا خانمی که به دلیل خونریزی ماهیانه شدید مراجعه کرده است و در آزمایشات پانسیتوپنی دارد.
شکل کمتر شایع و بدتر این است که دو ردهی پلاکت و گلبول قرمز کم باشند؛ اما گلبولهای سفید طبیعی تا افزایشیافته باشند.
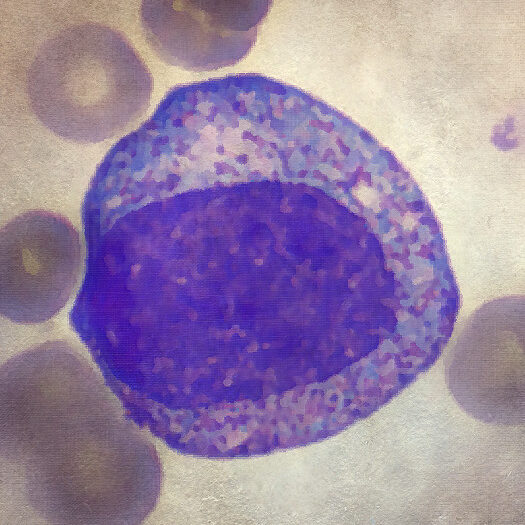
چه سلولهایی در لام خون محیطی ببینیم که به APL شک کنیم؟ در خون محیطی به دنبال پرومیلوسیت میگردیم.
درمان را پس از رؤیت پرومیلوسیت در لام خون محیطی آغاز میکنیم. میدانیم که در مغز استخوان، برای تبدیل شدن یک سلول به نوتروفیل بالغ، یک سری مراحل از میلوبلاست (Myeloblast) تا سلول باند (Band Cell) و پس از آن نوتروفیل بالغ طی میشود.
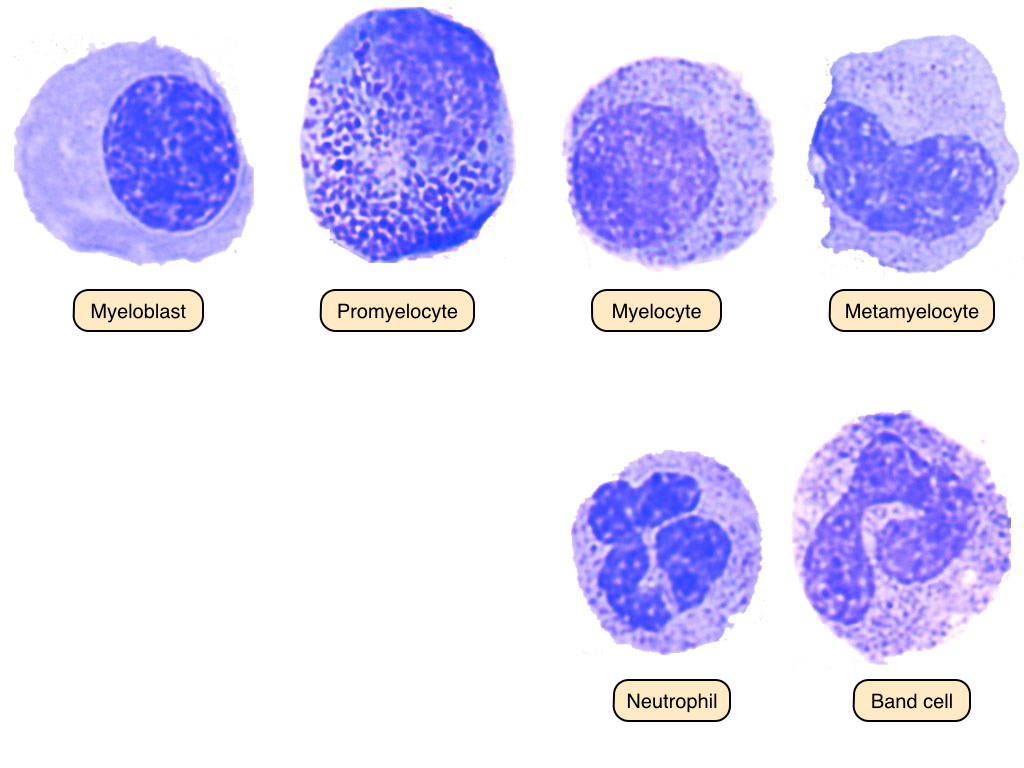
در هر مرحله یک سری اتفاق میافتد. میلوبلاست (سلول جوان) قرار است تبدیل به میلوسیت (سلول بالغ) شود و از سلول بالغ، نوتروفیل حاصل شود. مرحلهی قبل از به وجود آمدن میلوسیت، پرومیلوسیت (Promyelocyte) است. پیشوند Pro به معنای پیش و قبل است. در این مرحله سلول گرانولهای درشت دارد.
شبیه به اینکه یک خوشه انگور بنفش را دانهدانه کرده و درون سیتوپلاسم این سلول پخش کردهایم. دقت کنیم که این دانهها روی هسته نیز ممکن است قرار بگیرند. مثل بازوفیل، در پرومیلوسیت نیز گرانولها به حریم هسته احترام نمیگذارند و باعث میشوند هسته را کمی تار ببینیم.
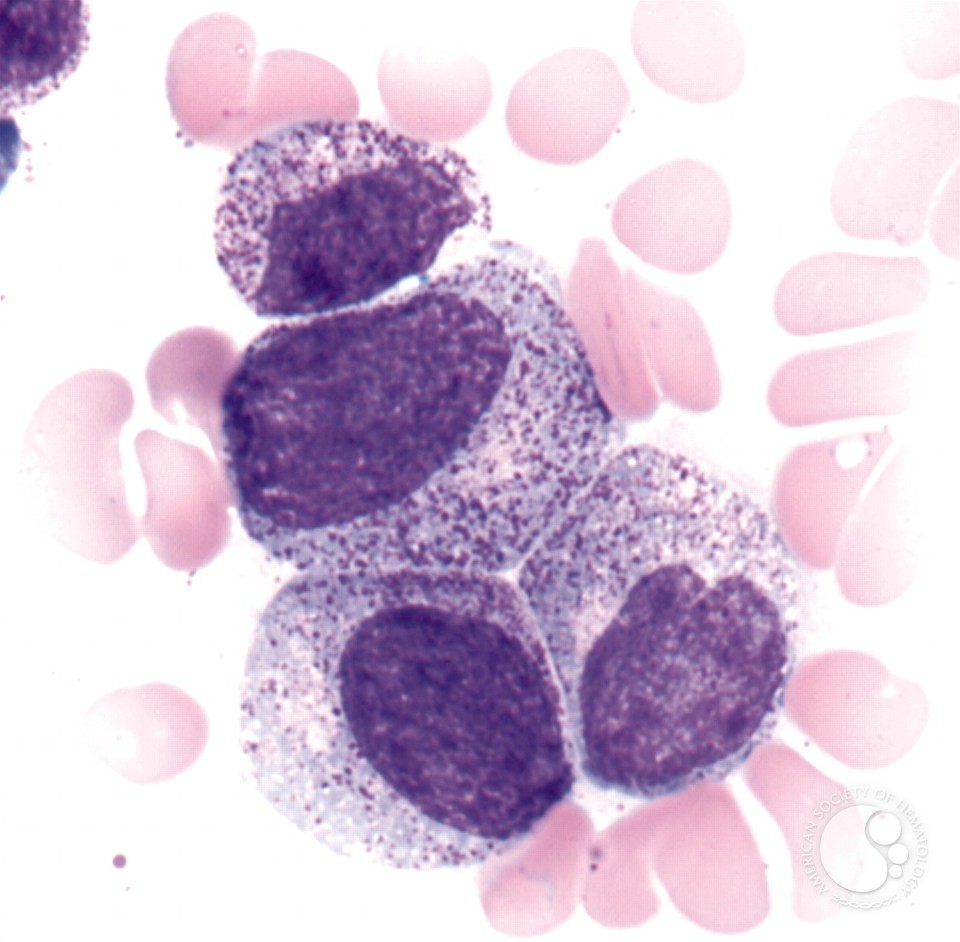
پس ویژگی پرومیلوسیت دیدن گرانولهای درشت است. این سلول معمولاً بزرگ بوده و چون سلولی نابالغ است، مادهی ژنتیکی داخل هسته، حالت فشرده ندارد و اصطلاحاً میگوییم که Fine Chromatin دارد. هستکها را نیز میتوانیم در هستهاش ببینیم.
این گرانولهای داخل سیتوپلاسم ممکن است به هم متصل شده و یک سری جسم میلهای شکل در سیتوپلاسم به نام Auer Rod به وجود بیاورند. این اجسام در اصل لیزوزومهای متصلشده هستند.
جان آئور (John Auer)، فیزیولوژیست برجستهی آمریکایی، از اولین افرادی – نه اولین فردی – بود که این اجسام را توصیف کرد و نام او روی این اجسام باقی مانده است. کمتر پیش میآید که ما بر اساس مورفولوژی تشخیص را قطعی کنیم؛ اما دیدن میلههای آئور، تقریباً وجود AML را قطعی میکند.

1948 – 1875

ممکن است در یک سلول فقط یک جسم آئور ببینم یا اینکه چند عدد از آنها همانند یک دسته هیزم کنار هم قرار بگیرند. در این حالت به سلول Faggot Cell میگویند. ریشهی کلمهی فاگوت به یک دسته چوب و هیزم برمیگردد. البته امروزه لغت فاگوت معنای توهینآمیزی در زبان عامیانه دارد و برای توهین کردن و مورد تمسخر قرار دادن پسران و مردان همجنسگرا به کار برده میشود.
احتمالاً ریشه این کلمه به این موضوع برمیگردد که در قرن شانزدهم زنان پیر بیوه را که با جمعکردن هیزم به گذران زندگی میپرداختند، Faggot-Gatherer (هیزم جمعکن) خطاب میکردند. البته این لفظ در همان زمان نیز معنای منفی داشت. پس دیدن Faggot Cell نیز به تشخیص کمک میکند. این سلولها در درون خود چند عدد Auer Rod دارند.

یک سلول دیگر نیز در AML-M3 ممکن است دیده شود که نام جا افتاده برای آن، Buttock Cell است. پرومیلوسیت معمولاً هستهای نسبتاً گرد دارد. گاهی این هسته یک شکاف پیدا میکند (Cleaved) و دو قسمتی میشود. برخی این شکل را شبیه به دو قسمت باسن دیده و برای همین به آن Buttock Cell میگویند. البته این نوع هستههای شکافدار مختص APL نیست و در سرطانهای دیگری نیز داده میشود.
این نوع سلول با هستهی شکافدار در نوعی از APL دیده شده که به آن Hypogranular Variant یا Microgranular Variant میگویند.
این سلولها، آن گرانولهای درشت بنفش رنگ را ندارند (نه اینکه اصلاً گرانول نداشته باشند. گرانولهای درشتی را که در بالا توصیف شد، نمیبینیم). به جایش هستهی دو لبه دارند که شبیه به دامبل، کلیه و یا باسن است که صرفاً اسم Buttock Cell رایج شده و Dumbbell Cell و Reniform Cell نداریم. البته به آن نام Bi-lobed Cell را هم دادهاند که در متون رسمی بتوانند نامش را با شرم کمتری بنویسند.
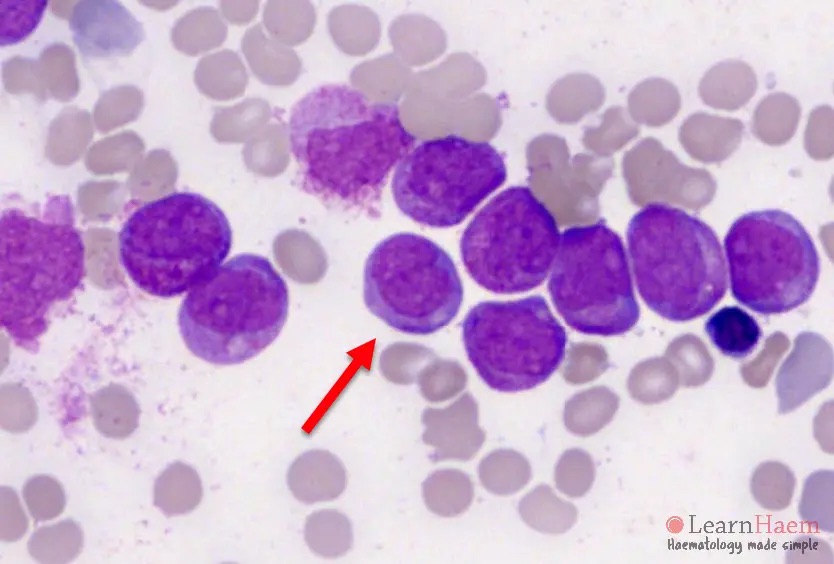
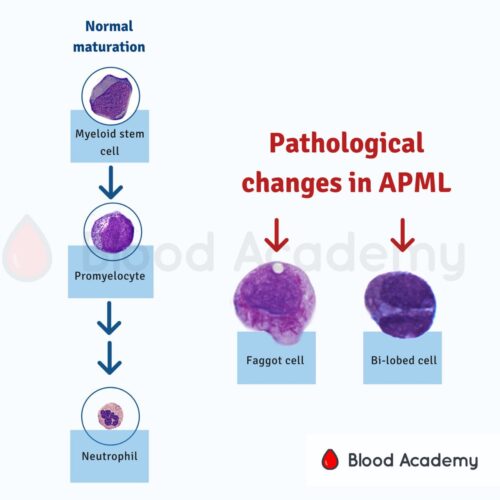
همچنین به این نکته دقت کنیم که در لام خون محیطی این افراد، نوتروفیل بالغ یا باند نمیبینیم. زیرا در سیر به وجود آمدن این سرطان، همانند خیلی از سرطانهای دیگر، یک توقف در تمایز (Maturation Arrest) داریم و سلول از پرومیلوسیت جلوتر نمیرود.
درمان دارویی
APL به دلیل میزان مرگ و میر بالا یکی از اورژانسهای پزشکی است. گایدلاینهای معتبر به صراحت اعلام کردهاند که درمان را بر اساس ویژگیهای بالینی و سیتولوژیک (لام خون محیطی) فوراً شروع کنیم و منتظر تأییدیه قطعی و تستهای ژنتیکی نباشیم.
در آنکولوژی، بدون داشتن نمونه و جواب پاتولوژی، معمولاً درمان شروع نمیشود. موقعیتهای انگشتشماری است که این قانون را زیر پا میگذاریم. یکی از آنها، AML-M3 است.
بعد از معرفی داروی All-trans-retinoic acid یا ATRA پیشآگهی APL به طرز چشمگیری بهتر شد. این دارو با اثر بر گیرندهی رتینوئیک اسید سبب متمایز شدن پرومیلوسیتها شده و آنها را به ردهی میلوسیت تبدیل میکند و ریسک DIC در بیماران کمتر میشود – زیرا که گرانولهای پرومیلوسیت بودند که DIC میدادند.
اگر بعد از شروع درمان با ATRA تشخیص به تأیید نرسید، آن را قطع و درمان را برای دیگر انواع AML شروع میکنیم.
با تجویز ATRA ریسک DIC کمتر میشود ولی ممکن است عارضه دیگری به اسم APL (Differentiation) Syndrome یا سندرم تمایز ایجاد شود. این سندرم در نتیجهی چسبیدن (Adhesion) سلولهای تمایز یافته به اِندوتلیوم عروق ریه ایجاد میشود و تابلوی بالینی شبیه Acute Respiratory Distress Syndrome یا ARDS ایجاد میکند. ظرف سه هفته از شروع درمان، بیماران دچار تب، احتباس مایع (و در نتیجه افزایش وزن)، تنگی نفس، درد قفسه سینه، ارتشاح (Infiltration) ریوی و هیپوکسمی میشوند. در گرافی شواهد افیوژن پلورال و پریکاردیال را میبینیم.
درمان APL Syndrome شامل کورتیکواستروئید (Dexamethasone به صورت ۱۰ میلیگرم وریدی هر ۱۲ ساعت برای سه روز)، درمان کاهندهی تعداد سلولها (Cytoreduction) و اقدامات حمایتی است.
در بیماران با APL Syndrome شدید (شامل آنهایی که دچار نارسایی کلیوی میشوند یا به دلیل دیسترس تنفسی نیازمند مراقبت ویژه هستند)، ATRA را موقتاً قطع میکنیم.
برای شروع درمان بیماران را به دو دسته Low-to-Intermediate Risk و High Risk تقسیمبندی میکنیم: دستهبندی بر اساس تعداد گلبولهای سفید (WBC count) انجام میشود. بیمار دسته Low-to-Intermediate تعداد گلبول سفید کمتر از ۱۰۰۰۰ در هر میکرولیتر دارند و بیماران High-risk تعداد گلبول سفید بالای ۱۰۰۰۰ در هر میکرولیتر دارند. بیماران High-risk احتمال بالایی برای خونریزی آلوئولار (Alveolar hemorrhage) و خونریزی مغزی دارند و باید بیشتر مراقبشان باشیم.
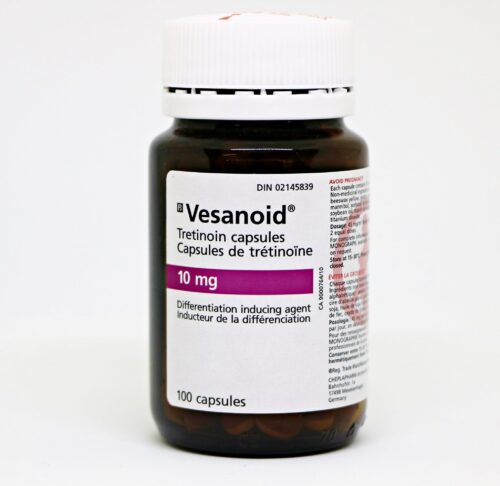
دوز ATRA معمولاً ۴۵ میلیگرم به ازای هر متر مربع از سطح بدن است. کپسولهای ATRA موجود در ایران ۱۰ میلیگرمی است. نام تجاری رایج آن Vesanoid است (برای شرکت Roche). در یک فرد با جثه معمولی تعداد ۸ کپسول کافی است. میتواند هر ۱۵ تا ۳۰ دقیقه یک کپسول مصرف کند یا اینکه هر ۱۲ ساعت ۴ کپسول یکجا مصرف کند. AML-M3 اصلاً مقاوم به درمان نیست و تقریباً همیشه با شروع این درمان به Remission میرود. این درمان اورژانسی است و با همین درمان میتوان جان بیمار را حفظ کرد.
رویکرد درمانی در بیماران High-risk کمی متفاوت است. در این دسته باید سریعاً درمان Cytoreduction را شروع کنیم چرا که بعد از شروع ATRA ریسک بالایی برای APL syndrome دارند و در معرض Induction Death و همچنین خونریزی ناشی از DIC هستند. البته که معمولاً درمان Cytoreduction توسط یک هماتولوژیست انجام میشود و ما نیز در این درس از توضیح داروهای آن (Daunrubicin و Cytarabine) خودداری میکنیم.
اما راه جایگزین نیز معرفی خواهیم کرد: برخی اساتید صاحب نظر برای افراد High-risk یا کسانی که با شروع درمان تعداد لکوسیتهایشان به بالای ۱۰۰۰۰ در هر میکرولیتر میرسد، برای پیشگیری از APL Syndrome از همان ابتدا کورتیکواستروئیدها را به درمان اولیه اضافه میکنند.
درمان حمایتی
پلاکت هدف در بیماران AML بالای ۳۰۰۰۰ پلاکت در هر میکرولیتر است ولی در APL هدف ما رساندن پلاکت به بالای ۵۰۰۰۰ در هر میکرولیتر است. همچنین سطح فیبرینوژن باید به بالای ۱۵۰ میلیگرم در دسیلیتر برسد.
در این بیماران پارامترهای انعقادی مثل فیبرینوژن، D-dimer، PT، aPTT و میزان پلاکتها به دقت مانیتور میشود.
پس انواع فرآوردههای پلاکت، (Fresh Frozen Plasma (FFP و Cryoprecipitate با هدف رساندن پلاکت بالای ۵۰۰۰۰ در هر میکرولیتر و فیبرینوژن بالای ۱۵۰ میلیگرم در دسیلیتر داده میشود.
نبایدها در فرد با AML-M3
برای Cytoreduction هرگز فرد با AML-M3 را Leukapheresis نکنید. لکوفورز در اینها ممنوع است چرا که ریسک خونریزی به علت Disseminated Intravascular Coagulation یا DIC را بالا میبرد.
هرگز برای بیمار با AML-M3، کاتتر ورید مرکزی (Central Venous Catheter) تعبیه نکنید. تحت هیچ شرایطی بیمار را Lumbar Puncture نکنید. در بیماران APL انجام این اقدامات تهاجمی قبل و حین درمان Induction ممنوع است.
پیام درس
هنگامی که یک فرد با پانسیتوپنی به همراه خونریزی یا ترومبوز مراجعه کرد، به AML-M3 شک میکنیم. شکل کمتر شایع و بدتر این است که دو ردهی پلاکت و گلبول قرمز کم باشند؛ اما گلبولهای سفید طبیعی تا افزایشیافته باشند. چه کار بکنیم؟ دیدن لام خون محیطی و به دنبال پرومیلوسیت گشتن. در صورت دیدن پرومیلوسیت، پس از فرستادن یا کنار گذاشتن نمونه خون محیطی جهت PCR برای PML-RARA:
۱. شروع ATRA به صورت اورژانسی. باز هم تأکید میکنیم که برای شروع ATRA نیازی به تأیید تشخیص از طریق پاتولوژی و نمونه مغز استخوان نیست. یک فرد با جثه متوسط، ۸ کپسول آترا (با نام تجاری وسانوئید) نیاز دارد که هر ۱۵ دقیقه یکی را بخورد.
۲. تزریق فرآوردهها شامل پلاکت، FFP و Cryoprecipitate با هدف پلاکت بالای ۵۰۰۰۰ در هر میکرولیتر و فیبرینوژن بالای ۱۵۰ میلیگرم در دسیلیتر.
۳. میتوانیم برای جلوگیری از APL Syndrome که تابلویی شبیه ARDS دارد، در افراد با WBC count بالای ۱۰۰۰۰ دگزامتازون (۸ تا ۱۰ میلیگرم وریدی هر ۱۲ ساعت) تجویز کنیم.
۴. از اقدامات تهاجمی مثل LP و تعبیه CV Catheter خودداری کنیم.
تجربه شما
یکی از بیمارانی را که دیدهاید و AML-M3 داشت، تعریف کنید. چطور به تشخیص رسیدید/رسیدند؟ علامت اولیه چه بود؟ برایش چه کار انجام شد؟
برای امتیاز دهی به این مطلب، لطفا وارد شوید: برای ورود کلیک کنید



بیماری داشتیم که خانم جوانی بود با لکوستاز و درنهایت DIC و ICH و اکسپایر شد. بارها کد خورده بود به خاطر قلب جوان و سالمش برمیگشت. شب تلخی بود…