
یکی از نامهایی که این روزها زیاد میشنویم، داروهایی با پسوند گلیفلوزین هستند، مثل امپاگلیفلوزین و داپاگلیفلوزین.
این داروها انتقالدهندهی سدیم-گلوکز شمارهی ۲ (sodium-glucose co-transporter 2 or SGLT2) را در کلیه مهار میکنند. به همین دلیل به آنها SGLT2 inhibitors یا اختصاراً SGLT2i میگویند.
داروهایی که نه تنها جایگاه متفورمین در درمان دیابت را به خطر انداختهاند، بلکه در درمان نارسایی قلبی نیز نقش ویژهای دارند.
شاید هیچ اندوکرینولوژیستی فکرش را هم نمیکرد که روزی مهار SGLT2 در کلیه، یکی از اهداف درمانی اصلی در دیابت نوع دو شود.
در این نوشته میخواهیم در مورد این داروها، تاریخچه کشف، مکانیسم اثر، جایگاهشان در درمان دیابت و نارسایی قلبی و همچنین عوارض مهمی که دارند صحبت کنیم.
نحوه اثر مهارکننده SGLT2
بازجذب مواد در کلیه به دو صورت فعال (با مصرف انرژی به دست آمده از متابولیسم ATP) و غیرفعال (بدون مصرف انرژی) انجام میشود.
انرژیای که برای انتقال فعال استفاده میشود، میتواند به صورت مستقیم و با مصرف آدنوزین تری فسفات (ATP) به دست آید که به آن انتقال فعال اولیه (primary active transport) میگویند. پمپ سدیم پتاسیم (Na-K ATPase) بدین شکل عمل میکند.
انرژی لازم برای انجام بعضی انتقالهای فعال نیز به صورت غیر مستقیم، مثلاً از انرژی نهفته در شیب الکتروشیمیایی یک یون، به دست میآید. به این روش، انتقال فعال ثانویه (secondary active transport) میگویند. بازجذب گلوکز در توبول پروگزیمال (proximal tubule) با این مکانیسم و با کمک شیب الکتروشیمیایی سدیم است.
یعنی همزمان که سدیم در جهت شیب غلظت شیمیایی خود به وسیلهی پروتئین انتقالدهندهاش به داخل سلول منتقل میشود، از انرژی آزاد شده از این انتقال، گلوکز نیز منتقل میشود.
درست است که برای انتقال گلوکز به داخل سلول مستقیماً انرژی مصرف نمیشود، اما برای حفظ شیب غلظت سدیم، فعالیت پمپ سدیم پتاسیم که با مصرف ATP سدیم درون سلول را پایین نگاه میدارد، نیاز است (secondary to primary active transport of sodium).
به این پروتئین انتقالدهنده، sodium-glucose cotransporter یا SGLT میگویند که به صورت بسیار مؤثر، تقریباً همهی گلوکز فیلتر شده از گلومرول را بازجذب میکند. آمینواسیدها نیز به همین شیوه به درون سلول منتقل میشوند.
بعد از اینکه گلوکز و آمینو اسید با همانتقالی سدیم به درون سلول آمدند، به صورت غیر فعال و بر اساس شیب غلظت خود، به بافت بینابینی در کورتکس کلیه منتقل شده و از آنجا به شبکه مویرگی دور توبولی میروند و به جریان خون باز میگردند.
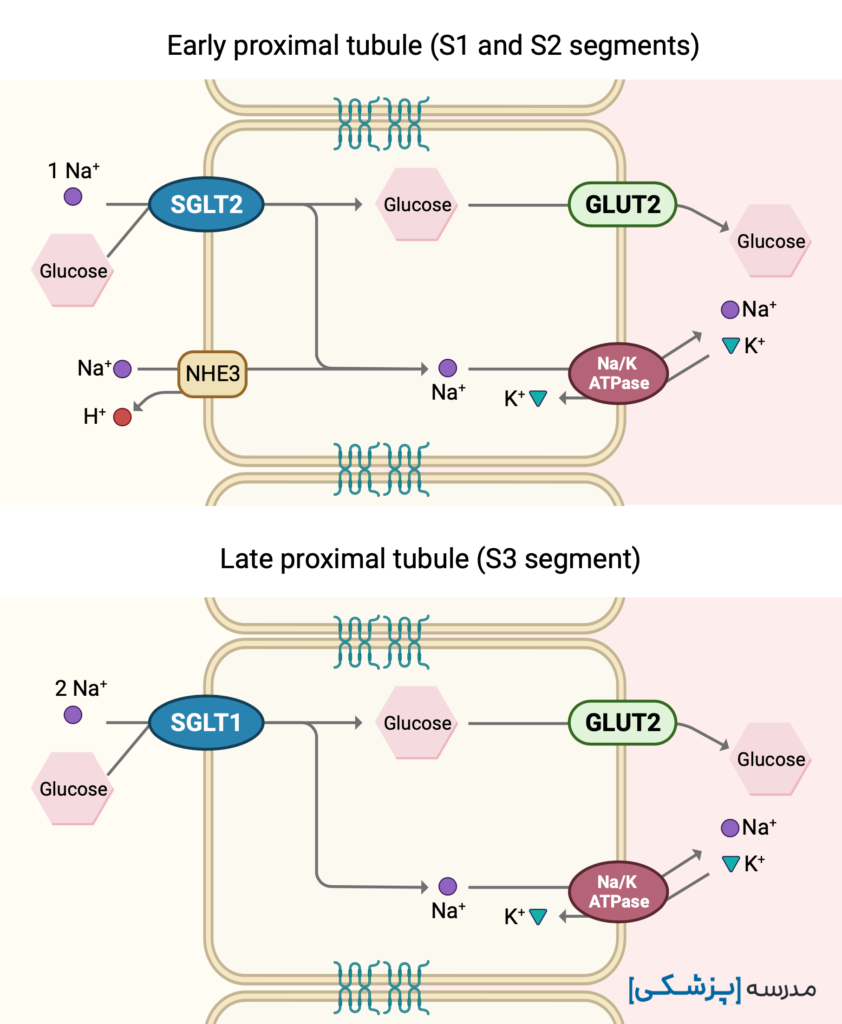
انتقالدهندههای سدیم-گلوکز دو نوع هستند (SGLT1 و SGLT2) و بر روی سطح لومینال اپیتلیوم توبول پروگزیمال (PCT) قرار دارند و گلوکز را با مصرف انرژی غیرمستقیمِ و بر خلاف شیب غلظت گلوکز، به درون سلول منتقل میکنند.
روزانه حدود ۱۲۰ تا ۱۸۰ گرم گلوکز از گلومرولها فیلتر میشود، اما فقط نیم گرم از آن از طریق ادرار دفع میشود.
حدود ۹۰٪ گلوکز فیلتر شده، خیلی زود توسط اولین سگمان توبول پروگزیمال به وسیلهی SGLT2 بازجذب میشود. ۱۰٪ باقیمانده نیز در دیگر سگمانهای توبول پروگزیمال و توسط SGLT1 بازجذب میشود.
قسمتهای مختلف توبول پروگزیمال
توبول پروگزیمال سه قسمت (segment) دارد. وقتی زیر میکروسکوپ نگاه کنیم، یک قسمت پیچخورده (convoluted) دارد و یک قسمت مستقیم (pars recta or straight).
اما مطالعات دقیقتر پروتئینها و عملکرد سلولها نشان داد که در اصل سه قسمت است:
- S1 که از گلومرول شروع شده و پیچخورده است.
- S2 که به شکل پیچخورده شروع شده و سپس مستقیم میشود.
- S3 که مستقیم است.
این سه قسمت ویژگیهای متفاوتی دارند.
در سطح بازولترال نیز، انتقال دهندههای گلوکز (glucose transporters یا GLUT) قرار گرفته دارند که گلوکز را بر اساس شیب غلظت از درون سلول به بافت بینابینی منتقل میکنند.
چرا دو نوع انتقالدهنده برای گلوکز داریم؟
اگر به قسمتهای مولکولی علاقه ندارید، میتوانید با خیال راحت از این قسمت بگذرید. این قسمت برای کسانی است که علاقهمند به دانستن جزئیات هستند.
SGLT در S1 و S2 که گلوکز فراوان است، ظرفیت بالا جهت انتقال دارد و تمایل نهچندان بالا برای گلوکز (high capacity, low affinity). زیرا آنقدر گلوکز زیاد است که نیاز ه تمایل بالا برای اتصال به گلوکز نیست. این SGLT همان SGLT2 است.
اما در S3 گلوکز کم است. بنابراین SGLT1 که در این ناحیه قرار دارد، تمایل بالا برای گلوکز داشته ولی در عوض ظرفیت بالایی ندارد (low-capacity, high affinity).
SGLT1 بر خلاف SGLT2 دو جایگاه برای سدیم داشته و از تمایل دو سدیم به شکل همزمان برای آوردن گلوکز به داخل استفاده میکند که بتواند این گلوکز کم را نیز به درون سلول آورد. SGLT1 در روده نیز قرار دارد.
چه وقتی گلوکز در ادرار میآید؟ (پدیده سرریز یا Splay Phenomenon)
حداکثر توان این SGLT چقدر است؟ حداکثر (maximum) چقدر گلوکوز را میتوانند منتقل (transport) کند؟ به این مفهوم transport maximum یا Tm میگویند.
مطالعات نشان داده که برای گلوکز در حدود ۳۷۵ میلیگرم در دقیقه است. یعنی مجموع SGLTها میتوانند در هر دقیقه تا ۳۷۵ میلیگرم گلوکز را منتقل (بازجذب) کنند.
اگر GFR را معادل ۱۲۵ میلیلیتر در دقیقه در نظر بگیریم:
125 ml/min × glucose concentration = 375 mg/min
glucose concentration = 3 mg/mL or 300 mg/dL
پس طبق این محاسبه، انتظار نداریم تا زمانی که قند بیشتر از ۳۰۰ میلیگرم در دسیلیتر شود، آن را در ادرار ببینیم. اما واقعیت اینطور نیست.
در واقعیت وقتی قند بیشتر از ۱۸۰ الی ۲۰۰ میلیگرم در دسیلیتر میشود، آن را در ادرار میبینیم.
این فاصلهی بین واقعیت و Tm را Splay یا سرریز کردن مینامیم. علت ایجاد این پدیده، یکدست نبودن نفرونهاست (tubular heterogeneity).
یک نفرون با پروگزیمال توبول کوتاه و گلومرول بزرگ، آنقدر توان ندارد که وقتی گلوکز بیشتر از ۱۸۰ میلیگرم در دسیلیتر میشود، تمام آن را بازجذب کند. طول این نفرون کوتاه، کفاف نمیدهد. در نتیجه گلوکز را به داخل ادرار سرریز میکند.
به عبارت دیگر، ما میدانیم که همهی نفرونها تا قند ۱۸۰ میلیگرم در دسیلیتر را میتوانند بازجذب بکنند. اما این حداکثر توان نفرون نیست. برخی همینجا محدود میشوند و برخی تا قند ۳۷۵ میلیگرم در دسیلیتر، قندی دفع نمیکنند.
با توجه به اینکه Tm را قبلاً برای بیکربنات نیز توضیح دادهایم، شاید این سؤال برایتان ایجاد شود که پس چرا بیکربنات «پدیده سرریز » را ندارد؟ علتش احتمالاً این است که بیکربنات جذب نشده میتواند در هنله یا جلوتر جذب شود. اما گلوکز فقط و فقط در توبول پروگزیمال جذب میشود.
گلوکز به عنوان یک قند، منبع اصلی انرژی برای بدن است. افراد دیابتی، به دلیل نداشتن انسولین یا وجود مقاومت در برخی از بافتها به آن، در استفاده از گلوکز دچار مشکل هستند. در نتیجه قند خون آنها بالا میرود. این هایپرگلایسمی در بلندمدت فرد را مستعد ایجاد عوارض متعددی در تقریباً همهی اعضای بدن میکند.
برخی افراد که قندشان چندان بالا نیست، با رژیم غذایی و کاهش وزن و ورزش بیماری خود را کنترل میکنند.
برخی نیز برای نگهداشتن قند خونشان در محدودهی طبیعی، نیاز به دارو دارند. این داروها یا انسولین هستند، یا به نحوی ترشح انسولین را در بدن تحریک میکنند؛ اصطلاحاً مکانیسمی وابسته به انسولین دارند.
برخی نیز مانند SGLT2i با مکانیسمهایی مستقل از انسولین عمل میکنند. این دارو با مهار SGLT2 و افزایش دفع کلیوی گلوکز اثر کاهندهی قند خود را میگذارد.
اما همه میدانیم و شنیدهایم که این داروها نقش محافظتی برای کلیه و قلب نیز دارند، درواقع renoprotective و cardioprotective هستند. این اثرات چگونه حاصل میشوند؟
در ابتدا داستان کشفشان را مرور کنیم و سپس به سراغ اثرات آنها بر قند و قلب و کلیه خواهیم رفت.
داستان کشف SGLT2i
جی.آر.آر تالکین زمانی نوشت:

این دقیقاً همان چیزی است که برای مهارکنندههای انتقالدهنده سدیم-گلوکز شماره ۲ افتاد.
شما به این محتوا دسترسی ندارید
برای مطالعه ادامه این مطلب نیازمند اشتراک ویژه مدرسه پزشکی هستید. خرید اشتراک از طریق صفحه شخصی امکانپذیر است.
ترتیبی که مدرسه پزشکی برای مطالعه مجموعه درسهای فارماکوپاتوفیزیولوژی کلیه پیشنهاد میدهد، به صورت زیر است:
برای امتیاز دهی به این مطلب، لطفا وارد شوید: برای ورود کلیک کنید



